石 河 子 大 学 讲 稿
课程名称 实验化学B-2
任课班级 2008级临床(3,7,8)班
任课教师 薛 梅
化工 学院 化学 系 公共有机 课程组
二OO八----二OO九学年第二学期
实验二 常压蒸馏及沸点测定
一、实验目的
1、掌握常量法和微量法测定沸点的原理和方法。
2、了解测定沸点的意义。
二、实验原理
1、什么是沸点?
2、沸点与外界压力的关系?
T0= t-(0.030+0.00011t)ΔP
式中: T0:标准状态下的沸点
t:实测沸点
ΔP:测定时的大气压与标准大气压之差
3、什么是蒸馏?
将液体物质加热到沸腾变成蒸气,再将蒸气冷凝为液体这两个过程的联合操作称为蒸馏。在常压下进行的蒸馏,称为常压蒸馏或普通蒸馏。
4、蒸馏的用途:
(1)分离液体混合物,但只有当混合物中各成分的沸点间有较大的差异(30℃以上)时才能有效地进行分离;
(2)测定化合物的沸点(常量法);
(3)提纯液体及低熔点固体,以除去不挥发的杂质;
(4)回收溶剂,或浓缩溶液。
5、蒸馏中须注意:
(1)为了消除在蒸馏过程中的过热现象和保证沸腾的平稳进行,以免液体突然暴沸,冲进冷凝管或冲出瓶外,造成损害甚至酿成火灾事故,加入沸石在加热前常加入几粒素瓷片或。由于它们表面疏松多孔,吸附有空气,可成为液体的气化中心,避免液体暴沸,这些物质又叫止暴剂。若加热后发现没有加止暴剂或原有的止暴剂失效时,必须先移去热源,待液体冷却后,再补加止暴剂。如中途停止蒸馏,切记在重新加热前补加新的止暴剂。
6、测定沸点的方法:蒸馏法也叫常量法,此法样品用量较多,要10ml以上。
微量法。
三、仪器和试剂
仪器 60ml蒸馏烧瓶、直形冷凝管、100℃温度计、接液管、50ml锥形瓶、50ml量筒、玻璃漏斗、沸石、打孔器。
试剂 酒精
四、实验步骤
(一)常量法
1、蒸馏装置
实验室的蒸馏装置主要包括下列三个部分:
(1)蒸馏烧瓶 一般使蒸馏物的体积不超过蒸馏瓶容积的2/3,也不少于1/3。温度计通过单孔塞插入瓶颈中央,其水银球的上缘恰好与蒸馏烧瓶支管接口的下缘在同一水平线上,这样才能保证在蒸馏时水银球完全被蒸气所包围,以便正确测出气液
平衡时蒸气的温度。

(2)冷凝管 液体沸点高于130℃的用空气冷凝管,低于130℃的用水冷凝管,一般用直形冷凝管。若液体沸点很低,则要用蛇形冷凝管。用水冷凝管时,其外套中通水,冷凝水从下口进入,上口流出,上端的出水口应向上,以保证套管中充满水。
(3)接收器 常由接液管和三角烧瓶或圆底烧瓶构成,两者之间不可用塞子塞住,应与外界大气相通,避免造成封闭体系。
2、蒸馏操作
仪器的安装顺序应从热源开始[1],自下而上,从左到右或从右到左,分别用铁架台、铁圈、铁夹予以固定,见图2—13。整个装置要求准确端正,横平竖直,无论从正面或侧面观察,全套仪器的轴线都要在同一平面内,所有铁夹和铁架台都应整齐地放在仪器的背面。
将30ml不纯乙醇通过玻璃漏斗加入蒸馏瓶中,注意勿使液体从支管流出。加入2~3粒沸石,塞好装有温度计的塞子,通入冷凝水[2],然后用水浴加热。开始火焰可稍大些[3],并注意观察蒸馏瓶中的现象和温度计读数的变化。当瓶内液体开始沸腾时,蒸气前沿逐渐上升,待达到温度计水银球时,温度计读数急剧上升,这时应适当调小火焰,控制馏出的液滴以每秒钟1~2滴为宜[4]。在蒸馏过程中,应使温度计水银球处于被冷凝液滴包裹状态,此时温度计的读数即为液体与蒸气平衡时的温度,也就是馏出液的沸点。当温度计读数恒定时,换一个已称量过的干燥的锥形瓶作接收器[5],收集馏分。当瓶内只剩下少量(约0.5~1ml)液体时,若维持原来的加热速度,温度计读数会突然下降,即可停止蒸馏。即使杂质很少,也不应将瓶内液体完全蒸干,以免发生意外。称量所收集馏分的重量或量其体积,并计算回收率。
蒸馏结束,先停止加热,后停止通水,拆卸仪器顺序与装配时相反。
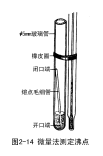
(二)微量法
微量法测定沸点采用图2-14所示的微量沸点管进行测定。先在外管中加入待测液体2-5滴,使液柱高约1cm,再放入内管。用橡皮圈把沸点管固定于温度计水银球旁,见图2-14。将温度计插入装有浴液的提勒(Thiele)管中(操作方法看熔点测定部分)。加热,由于气体膨胀,内管中有断断续续的小气泡冒出,到达样品的沸点时,将出现一连串的小气泡,此时停止加热,浴液温度自
 行下降,气泡逸出的速度逐渐减慢。仔细观察,最后
行下降,气泡逸出的速度逐渐减慢。仔细观察,最后
一个气泡出现而刚欲缩回至内管的瞬间,温度计所示
温度为毛细管内液体的蒸气压和大气压平衡时的温度,
亦即该液体的沸点[6]。再重复一次。纯净样品两次测
得的沸点差一般不超过1℃。
五、思考题
1、什么叫沸点?液体的沸点和大气压有什么关系?文献上记载的某物质的沸点是否即为你测得的沸点?
2、试述下列因素对常压蒸馏中测得的沸点的影响。
(1)温度控制不好,蒸出速度太快。
(2)温度计水银球上缘高于或低于蒸馏烧瓶支管下缘的水平线。
3、沸石的作用是什么?加热后才发现未加沸石时,应如何处理才安全?当重新进行蒸馏时,用过的沸石能否继续使用?
4、当加热后有馏液出来时,才发现冷凝管未通水,应如何处理?为什么?
5、如果液体具有恒定的沸点,那么能否认为它是单纯物质?
实验四 熔点测定及温度计校正
一、实验目的
1、掌握测定熔点的方法。
2、了解熔点测定的意义。
二、实验原理
1、什么是熔点?
通常把纯粹的固态物质受热熔化时的温度视为该物质的熔点。但是,严格的定义应该是,在标准大气压下物质的固态与液态成平衡时的温度。
2、熔点测定的意义?
利用熔点测定可以判断被测物质的纯度。
每种结晶的有机化合物,都有特定的分子间作用力,所以每种结晶的有机化合物都有特定的熔点。一个纯化合物从始熔到全熔的温度范围称为熔距(熔点范围或熔程),一般为0.5~1℃。若含有杂质则熔点下降,熔距增大。大多数有机化合物的熔点都在300℃以下,较易测定。若有两种物质A和B的熔点是相同的,可用混合熔点法检验A和B是否为同一种物质。若A和B不为同一物质,其混合物的熔点比各自的熔点降低很多,且熔距增大。
三、仪器和试剂
仪器 提勒管、温度计(150℃)、毛细管(内径1~1.5mm长15cm)、长玻璃管
(70~80cm)、玻璃钉、白瓷板、砂轮、小胶圈。
试剂 液体石蜡、苯甲酸、尿素、苯甲酸和尿素的混合物、未知样品。
四、实验步骤
1、毛细管法测定熔点
(1)熔点管的制备 见第一部分简单的玻璃工操作中拉制熔点管部分。
(2)样品的装填:装填样品的高度2~3mm
(3)测定熔点的装置 毛细管法测定熔点的装置有多种,常用的有两种。
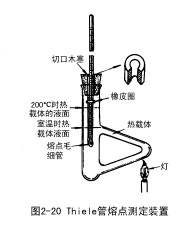
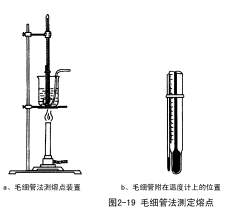
第一种装置 如图2-19(a)所示
第二种装置 如图2-20所示。将提勒(Thiele)管(又叫b形管或熔点测定管)夹在铁架台上,加入浴液至稍高于上侧管口上沿,再将配有温度计的缺口软木塞插入浴液中,使温度计水银球恰在提勒管的两侧管中部。毛细管如同前法固定在温度计旁。在图示部位加热。这种装置测定熔点的优点是管内液体因温度差而发生对流作用,省去了人工搅拌的麻烦。
浴液的选择:样品熔点在220℃以下的用液体石蜡或浓硫酸作浴液。
液体石蜡较安全,但易变黄;浓硫酸价廉,易传热,但腐蚀性强,
有机化合物与其接触,硫酸的颜色会变黑,妨碍观察,故装填样品时,沾在管外的样品
必须擦去。白矿油是碳原子数比液体石蜡多的烃,可加热到280℃不变色。还可用植物油、硫酸与硫酸钾的混合物、磷酸、甘油、硅油等作浴液。熔点测定完毕,浴液应倒回瓶中[5]。
(4)测定熔点 熔点测定的关键操作之一就是控制加热速度,使热能透过毛细管,
样品受热熔化,令熔化温度与温度计所示温度一致。此时应该特别注意温度的上升和毛细管中样品的变化情况。样品将依次出现“发毛”、“收缩”、“液滴”、“澄清”等现象,发毛和收缩以及形成软质柱状物而无液化现象都不是“始熔”,只有当出现液滴(塌落,有液相产生)时才是“始熔”,全部样品变成透明澄清液体时为“全熔”见图2-21。记录“始熔”与“全熔”时温度计上所示的温度,其差值即为该化合物的熔程。例如某一化合物在112℃时开始收缩,113℃时有液滴出现,在114℃时全部成为透明液体,应记录为:熔点113~114℃。熔点测定至少要有两次重复数据。熔点管不能重复使用,每一次测定,都必须用新的熔点管另装样品,。
(5)实验内容 用第二种装置及液体石蜡为浴液,按前述方法测定尿素、苯甲酸、尿素和苯甲酸的混合物及未知样品的熔点。
五、思考题
1、三个瓶子中分别装有A、B、C三种白色结晶的有机固体,每一种都在149~150℃熔化。一种A与B的等量混合物在130~139℃熔化;一种A与C的等量混合物在149~150℃熔化。那么B与C的等量混合物在什么样的温度范围内熔化?你能说明A,B,C是同一种物质吗?
2、测定熔点时,若遇下列情况,将产生什么结果?
(1)熔点管壁太厚。
(2)熔点管底部未完全封闭,尚有一针孔。
(3)熔点管不洁净。
(4)样品未完全干燥或含有杂质。
(5)样品研得不细或装得不紧密。
(6)加热太快。
实验七 折光率测定
一、实验目的
1、掌握测定有机化合物折光率的方法。
2、了解测定折光率的意义及折光仪的构造。
二、实验原理
折光率是有机化合物的重要物理常数之一。其测定简单、方便且准确,作为液体有机化合物的纯度标准,比沸点更可靠。折光率可用以鉴定未知物,也可用于确定液体混合物的组成。
光线自第一种介质进入第二种介质时,由于两种介质不同,光线在两种介质中传播的速度也不同,所以光线就发生折射。入射光线与法线的夹角称为入射角i;折射光线与法线的夹角称为折射角γ,见图2-29。
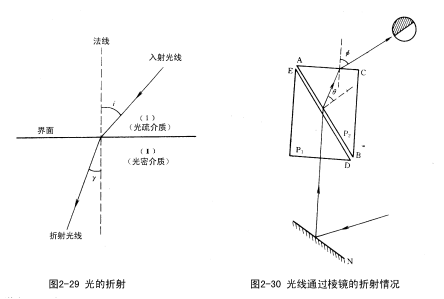
由实验证明入射角i的正弦与折射角γ的正弦之比为一常数,它等于光线在两种介质中的传播速度之比,即:
n =![]() n =
n =![]()
υ1代表光线在第一种介质中的传播速度,υ2代表光线在第二种介质中的传播速度,n是常数。通常以空气作为标准介质,当光线由空气进入另一种物质,所得的速度比即为该物质的折光率。由于光线在空气中的速度比在液体中的速度大,故液体的折光率总是大于1。
当入射角接近或等于90°时,折射角就达到了最大折射角,此角称为临界角,用γ0表示,测定临界角就可算出折光率。
测定折光率的仪器叫折光仪。
物质的折光率不但与它的结构和测定时所用光线的波长有关,而且也受温度等因素的影响。所以折光率的表示须注明所用光线的波长和测定时的温度,常用n![]() 表示。D是以钠灯的D线(58930nm)作光源,t是测定时的温度。例如n
表示。D是以钠灯的D线(58930nm)作光源,t是测定时的温度。例如n![]() 表示20℃时,该物质对钠灯的D线的折光率。
表示20℃时,该物质对钠灯的D线的折光率。
通常温度升高1℃,液体化合物的折光率降低3.5![]() 10-4~5.5
10-4~5.5![]() 10-4。
10-4。
三、仪器和试剂
仪器 阿贝折光仪、擦镜纸、滤纸
试剂 重蒸馏水、丙酮、松节油、乙酸乙酯、葡萄糖溶液
四、实验步骤
1、Abbe折光仪的校正
用重蒸馏水校正,重蒸馏水的折光率(n![]() =1.3337,n
=1.3337,n![]() =1.3330,n
=1.3330,n![]() =1.3325)
=1.3325)
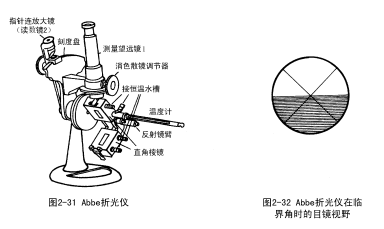
2、样品的测定
读数应保留小数点后四位,重复3~5次(误差不大于0.002)取平均值,即为所测物质的折光率。
如果在目镜中看不到半明半暗,而是畸形的,这是因为棱镜间未充满液体,若出现弧形光环,则可能是有光线未经过棱镜面而直接照射在聚光透镜上。若液体折光率不在1.3~1.7范围内,则Abbe折光仪不能测定,也调不到明暗分界线。
(3)测定完毕,打开棱镜,用擦镜纸擦去试样,并用丙酮或无水乙醇清洗棱镜,晾干后,棱镜之间垫一片擦镜纸,关紧棱镜。
(4)糖溶液浓度的测定 按上述方法测定糖溶液的浓度,读数时从读数镜视场左边所指示值读出,即为糖溶液含糖量浓度的百分比数。
3、Abbe折光仪的维护
(1)Abbe折光仪在使用前后,棱镜均需用丙酮或乙醚洗净,并干燥之,滴管或其它硬物均不得接触镜面,擦洗镜面时只能用丝巾或擦镜纸吸干液体,不能用力擦,以防损坏镜面。
(2)用完后,放出金属套中恒温水,拆下温度计并放在纸套筒中,将仪器擦净,放入箱中。
(3)折光仪不能放在日光直射或靠近热源的地方,以免样品迅速蒸发。仪器应避免强烈振动或撞击,以防光学零件损伤及影响精度。
(4)酸、碱等腐蚀性液体不得使用Abbe折光仪测其折光率,可用浸入式折光仪测定。
(5)折光仪不用时需放在木箱内,箱内应贮有干燥剂,木箱应放在干燥、空气流通的室内。
五、思考题
1、测定物质折光率的意义是什么?折光率随什么因素而改变?
2、假定松节油的折光率n![]() =1.4710,在25℃时折光率的近似值是多少?
=1.4710,在25℃时折光率的近似值是多少?
实验八 旋光度的测定
一、实验目的
1、熟悉旋光仪的基本原理。
2、掌握旋光度的测定方法。
二、实验原理
旋光度除与光学活性物质的本性有关外,还与光源的波长、温度、溶液的浓度(若为纯液体则为密度)、盛液管的长度(光线通过路径的长度)及测定时所用的溶剂有关,若将波长和温度固定,则:
α=[α]![]() ·l·C 或 [α]
·l·C 或 [α]![]() =
=![]()
α:旋光度,它与盛液管的长度l(以分米为单位)和溶液的浓度C(每毫升含的克数)成正比,[α]![]() 为比例常数,称为比旋光度。它是以钠光的D线为光源,20℃时[1],用1分米长的盛液管,测得每毫升含1克旋光性物质的溶液的旋光度。比旋光度是旋光性物质的物理常数之一,应写作[α]
为比例常数,称为比旋光度。它是以钠光的D线为光源,20℃时[1],用1分米长的盛液管,测得每毫升含1克旋光性物质的溶液的旋光度。比旋光度是旋光性物质的物理常数之一,应写作[α]![]() ,需注明所用溶剂。
,需注明所用溶剂。
测定旋光度的仪器叫旋光仪,见图2-35。
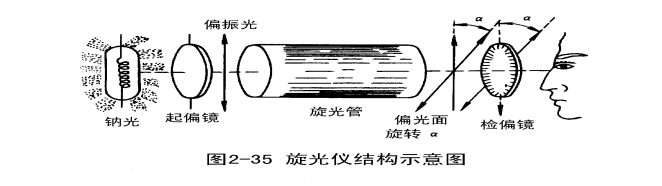
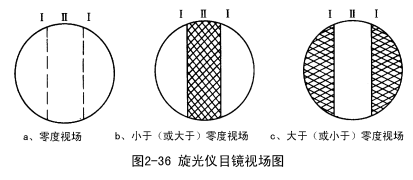
由于人的眼睛确定全黑暗或完全明亮的瞬间有些困难,因此在旋光仪里设了一种三分视场(也有二分视场的),见图2-36,以提高测量的准确度。
其原理如下:在起偏镜后的中部装一狭长的石英片,宽度约为视场的1/3,由于石英具有旋光性,故从石英片中透过的那一部分偏振光被旋转了个角度,此角称为半暗角,此时若:
1、检偏镜的光轴与起偏镜的光轴平行,则视场中间稍暗,两旁很亮。
2、检偏镜的光轴与石英片的光轴平行,则视场中间很亮,两旁稍暗。
3、检偏镜的光轴与起偏镜的光轴垂直,则视场中间稍亮,两旁很暗。
4、检偏镜的光轴与石英片的光轴垂直,则视场中间很暗,两旁稍亮。
5、检偏镜的光轴与半暗角的平分线垂直,则视场的中间部分光与两旁光的暗度相同,三分视场消失。此法确定零点最为灵敏,故将该视场称为“零度视场”。
6、检偏镜的光轴与半暗角的平分线平行,则视场的中间部分光与两旁光的亮度相同,整个视场很亮,故称亮视场。此时不利于判断三分视场是否消失,故不能以此角度作为测定旋光度的零点。
三、仪器和试剂
仪器 旋光仪
试剂 葡萄糖溶液、果糖溶液
四、实验步骤
1、旋光仪零点的测定
2、旋光度的测定
五、思考题
1、比旋光度[α]![]() 与旋光度α有何不同?
与旋光度α有何不同?
2、测葡萄糖溶液的旋光度,能否用新配制的?为什么?
3、在本实验中,为什么要用不同长度的盛液管测同一样品?
实验九 层析法
一、实验目的
学习柱层析、纸层析、薄层层析的原理及其方法。
二、实验原理
层析法(又称色谱法,色层法或层离法),是分离、提纯、鉴定有机化合物的重要方法。层析法于1903年提出,开始仅用于分离有色化合物,随着显色方法的引入,现己广泛应用于无色化合物的分离和鉴定。近年来层析法在化学、生物学、医学中都得到了普遍应用,它帮助解决了天然色素、蛋白质、氨基酸、生物代谢产物、激素和稀土元素等的分离和分析。
层析法是一种物理分离方法,其基本原理是利用混合物各组分在某一物质中吸附或溶解性能(即分配)的不同,或其它亲和作用性能的差异,使混合物溶液流经该种物质,进行反复的吸附或分配等作用,从而将各组分分开。流动的混合物溶液称为流动相,固定的物质称为固定相(可以是固体或液体)。
层析法按其两相所处的状态可分为液相层析和气相层析。前者以液体为流动相,后者以气体为流动相,按层析原理可分为吸附层析、分配层析、离子交换层析和凝胶层析;按操作形式又可分为柱层析、薄层层析、纸层析。
(一)柱层析
常用的有分配柱层析和吸附柱层析两类。
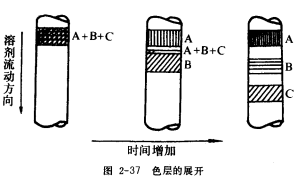
吸附柱层析通常在层析柱中填入表面积很大,经过活化的多孔性物质或粉末状固体吸附剂(如氧化铝或硅胶)作固定相,当混合物溶液从柱顶加入,流经吸附柱时,即被吸附在柱上端,当从柱顶加入溶剂(洗脱剂)洗脱时,各组分由于被吸附能力不同,而以不同的速度解析,随溶剂下移,经过反复吸附与解析,逐渐形成若干色带,见图2-37;继续加入溶剂洗脱,则吸附力最小的组分随溶剂首先流出,分别收集各组分,再逐个鉴定。柱上不显色的化合物可用紫外光照射后呈现的荧光来检查,或在洗脱时,分别收集一系列固定体积的流分,逐个鉴定,从柱内洗下来的物质组成可通过测定物理常数或光谱,并与己知物质比较,而予以确定。
分配柱层析常以硅胶、硅藻土和纤维素为支持剂,以其中吸收的液体作固定相,利用混合物组分在两种不相溶的液体中分配不同而得到了分离,相当于连续性溶剂萃取。
在进行吸附柱层析的实验中,有许多应注意的问题,它们能直接影响分离的效果。
1、吸附剂
常用吸附剂有氧化铝、硅胶、氟化硅多聚体、氧化镁,碳酸钙、淀粉及糖等。选择吸附剂的首要条件是与被吸附物无化学作用。吸附剂的吸附能力与其颗粒大小有关,颗粒太粗,流速快但分离效果不好;太细则流速慢。层析用的氧化铝有酸性、中性和碱性三种,分别用于分离酸性、中性和碱性物质。
化合物被吸附的能力与吸附剂和化合物的极性有关。极性吸附剂对极性强的化合物吸附力强,对极性弱的化合物吸附力弱。已经发现具有下列极性基因的化合物,在任一给定的吸附剂上的吸附力增加次序为:
2、溶剂
溶剂也可能吸附在固体上,与溶质竞争表面上的吸附位置。为了有效分离,洗脱剂极性必须明显小于混合物组分的极性,且能溶解组分。不同溶剂的洗脱力次序为:
己烷<CC14<甲苯<苯<CH2Cl2<CHC13<乙醚<乙酸乙酯<丙酮<丙醇<乙醇<甲醇<水
理想的溶剂应能使层带达到最大分离。选择最佳溶剂常须反复试验,为了提高溶剂之洗脱能力,也可选择混合溶剂。
3、装柱
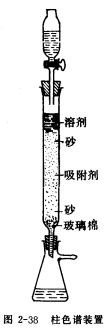
吸附柱层析的分离效果不仅依赖于吸附剂与洗脱溶剂的选择,而且与制成的层析柱有关,柱层析的装置见图2-38。柱的大小视处理量而定,一般吸附剂用量应为被分离样品的30-40倍,柱高和直径比为7.5:1。装柱可采用干法和湿法两种装柱方法。
(1)干法装柱 取少许脱脂棉(或玻璃棉)于干净的层析柱底部,轻轻塞紧,上盖一层0.5cm厚的石英砂(或用一张略小于柱内径的滤纸代替),关闭活塞,向柱中加入洗脱剂至柱高的3/4处,打开活塞,控制流速为1滴/秒,用一玻璃棒压实脱脂棉,以除去其中空气泡,然后通过一干燥玻璃漏斗慢慢加入吸附剂,同时轻轻敲击柱身下部,使填装紧密,加入吸附剂的高度应为柱径的10一15倍,最后使吸附剂表面水平,在其上加一滤纸,再加一层0.5cm厚的石英砂以保护柱顶表面。注意:溶剂液面不得低于柱顶表面。
(2)湿法装柱
其步骤与干法装柱基本相同,只是加入的吸附剂应在溶剂中制成糊状物之后,再加入层析柱中。
不论采用哪种方法装柱,都应注意将吸附剂装填紧密均匀,不要有裂缝或气泡,否则会影响分离效果。
(三)纸层析
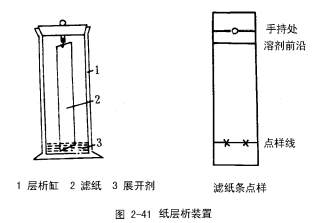
纸层析是一种液一液分配层折,以特制的滤纸为支持剂(或称载体)以滤纸纤维上吸附的水份为固定相,流动相是含有一定比例水的有机溶剂(常称展开剂),所需分离的样品点在滤纸条的近底部,滤纸再放入层析缸中,见图2-41。样品以下的部分浸在溶剂里,利用纸纤维的毛细作用,溶剂在含水的滤纸上移动,依据被分离物在两相分配系数的不同而达到分离目的。
与薄层层析相同,样品经纸层析后,被分离开的各种化合物可通过比较其Rf值来鉴定。
三、仪器和试剂
仪器 层析柱一支、接收瓶3个、烧杯一个、玻璃棒、滴管、小滤纸、层析槽、毛细管、回流装置(100ml短颈蒸馏烧瓶、球形冷凝管)、层析缸、抽滤装置、电吹风机、新华2号或1、3号滤纸。
试剂 乙醇(0.95)、层析用氧化铝(160-200目)、玻璃棒、石英砂、展开剂(环己烷:乙酸乙酯=9:1)、盐酸(0.19)、活性炭、沸石、氨基酸标准样品、展开剂(正丁醇:醋酸:水=4:1:5)、显色剂(茚三酮乙醇溶液)(10g.L-1)、0.01%荧光黄-亚甲基蓝乙醇溶液、苏丹Ⅲ苯溶液、偶氮苯苯溶液、苏丹Ⅲ和偶氮苯混合物苯溶液、头发。
四、实验步骤
(一)柱层析分离荧光黄和亚甲基蓝
1、装柱
按前述方法以氧化铝为吸附剂,95%乙醇为洗脱剂,进行湿法装柱。
2、加样
放出柱内溶剂,当溶剂液面降至几乎与氧化铝表面齐平时,关闭活塞。滴加试样溶液1ml于柱中(尽量不要碰到管壁上),打开活塞,当液面降至几乎与柱顶齐平时,关闭活塞,用吸管吸少量(0.5ml)乙醇(0.95)淋洗管壁上可能吸附的试样,如此连续2-3次,直至洗净为止。
3、洗脱,分离
向柱内缓缓加入乙醇(0.95)进行洗脱。当亚甲基蓝洗脱出柱时,更换接收瓶收集之,亚甲基蓝洗脱完后,再换另一接收瓶,用水作洗脱剂,将荧光黄洗出并收集之。
注意在整个洗脱过程中,应始终保持溶剂液面不得低于石英砂面;流速可控制在1-5滴/秒。
(三)纸层析法分离头发水解产物中的氨基酸
1、头发水解产物的制取
在100ml短颈烧瓶中加入0.5g头发、20ml盐酸(0.19)和2-3粒沸石,安装好回流装置,见图1-6,直接加热,回流约30分钟(火温不要太高,保持沸腾即可)。稍冷后在水解液中加入0.5g活性炭,煮沸5-10分钟,抽滤,滤液待用[2]。
2、纸层析
(1)点样 取层析滤纸一条[3],在其一端2-3cm处,用铅笔轻轻画一横线作为起点线,用毛细管分别沾取步骤1中的滤液和氨基酸标准品,轻轻点于起点线上(两样品应尽量分开,同时距滤纸左右边缘至少应1cm,以免产生边缘效应),若一次点样不够,可在干后再点1-2次,.每次点样后,样品扩散直径不应超过0.3cm。
(2)展开 展析缸中注入8ml展开剂(注意展开剂不要碰到容器壁上),将点样的滤纸悬挂在层析缸内密闭饱和10min[4],然后将点样端浸入展开剂约1cm(展开剂不得触及点样处),盖好盖令其展开,展开剂前沿上升至原点的距离不得小于10cm。
(3)显色 展开完毕,取出滤纸,立即用铅笔轻轻标记出展开剂前沿位置,用吹风机吹干后,喷洒显色剂(或将其浸入显色剂溶液内润湿),再以热风吹干至显色,用铅笔画出显色斑点的位置。
(4)计算各种氨基酸的Rf值,并与标准品Rf值比较。
五、思考题:
1、为什么极性大的组分要用极性大的溶剂洗脱?
2、层析柱中若留有空气或填装不匀,会怎样影响分离效果?
5、在一定操作条件下,为什么可用Rf值鉴别化合物?
6、含两个极性不同组分的样品,在一极性大的展开剂中展开时,可否预计两个组分展开后Rf值的大小,为什么?
7、层析滤纸被手汗污染,对层析结果有什么影响?
Chapter 16 Preparation of acetylsalicylic acid (esterification)
Goals
(1)Master the principle and method of synthesis of acetylsalicylic acid.
(2)Master the technique of recrystallization by using inflammable solvent.
Caution
Acetic anhydride is a corrosive lachrymator.
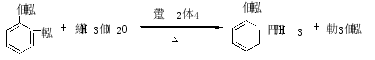
1、Principle
Nucleophilicity of phenolic hydroxy is weaker than that of alcoholic hydroxy,the usual phenolic hydroxy can be acylated by only stronger acylating agent,such as acyl halides or anhvdrides.
2、Reagents
salicylic acid l0g (0.0724mo1)
acetic anhydride l5g (14ml,0.147mol)
conc.H2SO4 5 drops (about 0.0047mol)
petro1eum ether(b.P.30℃~60℃)
3、Procedure
Place 10g(0.0724mo1)of dry salicylic acid and 15g(14m1,0.147mol)of acetic anhydride in a small conical flask,add 5 drops of concentrated sulphuric acid and start magnetic stirrer in order to secure thorough mixing. Warm on a water bath to about 50℃~60℃ stirring for about l5 minutes. Allow the mixture to cool and stir occasional1y. Add 150 ml of water, stir well and filter at the pump.Dissolve the solid in about 30 ml of hot ethanol and pour the solution into about 75 ml of warm water,if a solid separtes at this point,warm the mixture until solution is complete and then allow the clear solution to cool slowly.Beautiful needle-like crystals will separate. The yield is 11g(85%).The air-dried crude product may also be recrystallized from ether-light petroleum(b.p.30℃~60℃).
Acetylsalicylic acid decomposes when heated and does not possess a true,clear1y defined m.p. Decomposition points varying from l28℃ to l35℃ have been recorded,a value of l29℃~133℃ is obtained on an electric hot plate. Some decomposition may occur if the compound is recrystallized from a solvent of high boiling point or if the boiling period during recrystallization is unduly prolonged.
Physical constants of main Feagents and product
name | molecular weight | Physical state | density | Melting point (℃) | Boiling point(℃) | Solubility | ||
water | ethanol | ethyl ether | ||||||
Acetic anhydride | 102.09 | Colorless liquid | 1.0802 | -73.1 | 140 | 13.6 decom- pose | ∞ | ∞ |
Salicylic acid | 138.12 | White crystal | / | 157~159 | / | 0.22 | Easy solu-ble | Easy solu-ble |
Acetylsa-licylic acid | 180.16 | White crystal | / | 138~140 | / | 0.3325
| Very solu-ble | Easy solu-ble |
Notes
[1]Salicyh:acid require to be powdered before the use.
[2]Pay attention to moisture proof.
[3] Don’t use open fire during recrystallizing aspirin by using inf1ammab1e solvents.
4、Questions
(1)In this experiment,can you replace acetic anhydride with glacial acetic acid?
(2)Can acetylsalicylic acid recrystallize from water?
(3)Why do you use excess acetic anhydride?
实验十 模型作业
一、实验目的
1、加深对有机分子的立体概念,能把平面结构式转换成立体模型。
2、用立体概念理解分子平面图形的某些特性。
二、实验原理
三、模型
五种不同颜色具有适当孔眼的球、直棍、弯棍模型一套。
四、实验步骤
(一)构象
1、搭出乙烷和丁烷的分子模型
2、搭出环己烷分子的椅式和船式构象模型
3、搭出甲基环己烷的椅式构象模型,然后将其扭转成另一种椅式构象,观察a-或e-(特别是连甲基的)键有何变化?室温时的优势构象为哪种?
4、搭出1,4-二甲基环已烷的三种椅式构象模型,其相对稳定次序如何?室温时的优势构象为哪种?
5、搭出十氢化萘的两种构象模型,其中两个椅式环己烷各为何种稠合方式?室温时的优势构象为哪种?
(二)顺反异构
1、搭出两种不同构型的2-丁烯分子模型,观察两者能否重合?何者较稳定?
2、搭出1,3-丁二烯的两种构型的分子模型,分析为什么S-反式比S-顺式稳定?
3、搭出1,4-环己烷二羧酸的两种构型的分子模型,两者能否重合?何者较稳定?
4、搭出2-丁烯酸的两种构型的分子模型,两者能否重合?何者较稳定?
5、搭出1-氯-2-溴丙烯的两种构型的分子模型,两者能否重合?何者较稳定?
(三)旋光异构
1、用五种不同颜色的球搭出乳酸两种构型的分子模型,观察其是否有对称因素?两者互为什么异构体?能否重合?有无旋光性?
2、用五种颜色不同的球搭出2-羟基-3-氯丁二酸四种构型的分子模型,四者能否重合?互为什么异构体?
3、用四种不同颜色的球搭出酒石酸的分子模型,你能搭出几种?它们能否重合?互为什么异构体?何者为同一物质?何者为内消旋体?
4、用单键将两个苯环相连,再用两种不同颜色的大球代表硝基和羧基,分别接在相应的位置上,即可构成2,2’-二硝基-6,6’-二羧基联苯分子的模型。观察2,2’-和6,6’-位所连基团的作用力,两个苯环能否共平面?该化合物是否具有旋光性?为什么?
5、用三种不同颜色的球搭出2-甲基-4-溴-2,3-戊二烯的分子模型,观察两个π健能否共平面?该化合物有无对称因素?
(四)、糖的结构
1、搭出链状葡萄糖的分子模型,然后将其碳链竖向放置,令醛基向上,羟甲基向下,再向右翻平。旋转C5使其羟基与醛基碳靠近,将醛基碳(Sp2杂化)换成Sp3杂化碳,把C5羟基中的氢原子脱离氧原子而与醛基氧相连,其氧原子与醛基碳相连即构成环状葡萄糖的模型。观察它是α一型还是β一型?再将其扭转成环状葡萄糖的构象式模型,观察β一型中较大的基团是否都在e键上?在α一型中除C1上的基团外,其余各碳原子上的较大基团是否也在e健上?
2、仿照上法搭出链状果糖的分子模型,然后将其碳链竖向放置,令羰基向上,再向右翻平。旋转C5或C6使其羟基与羰基碳靠近,将羰基碳换成Sp3杂化碳,把C5或C6羟基中的氢原子脱离氧原子,而与羰基氧相连,其氧原子与羰基碳相连,即构成呋喃果糖或吡喃果糖的模型。它是α一型还是β一型?
五、思考题
1、为什么链状烷烃的碳链以锯齿形排列时最稳定?
2、环丁烷或环戊烷的碳原子能否在同一平面上?它们各以何种构象存在?
3、写出1,4-二甲基环已烷所有的椅式构象式。
4、指出下列化合物有无顺反异构体?若有,试用顺/反和Z/E两种方法分别加以命名。
(1)1一苯基一1一丙烯 (2)1,1,2一三甲基环已烷
(3)2一甲基一1一丁烯 (4)1一氯一1,2一二溴乙烯
(5)2,3一二氯一2一丁烯
5、试写出L-一(+)果糖和L一(一)葡萄糖的链状费歇尔投影式。
1